
By Dr. Colin Michie FRCPCH University of Central Lancashire.
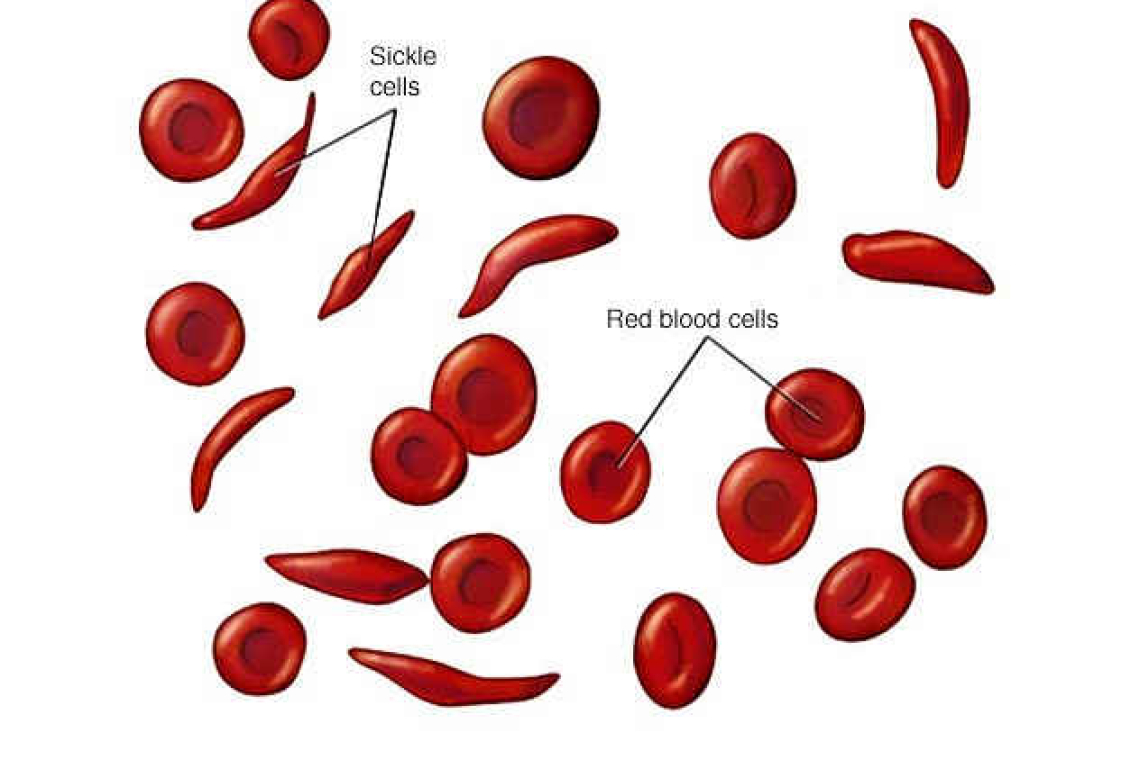
Our neighbours gifted a home DNA test kit to their daughter for her 18th birthday. This showed unexpectedly that she had sickle cell trait. Within days, grandparents, uncles and aunts began to visit. For several weeks, there was great anxiety while everyone was checked. No one in the family knew much about sickle-cell anaemia. This condition is caused by an inherited genetic change. Although the sickle cell mutation is simple, its impacts may be widespread.
That DNA change can cause packed haemoglobin molecules to form sheets, or become lumpy, inside red cells. The red cells then deform and burst more readily, releasing iron and haemoglobin into the bloodstream. This causes anaemia, a reduction in the numbers of red cells. The process can often make blood vessel walls become inflamed and sticky: it can block small blood vessels. The results may include pain lasting for several days; there might be pneumonia; there may be a large loss of red cells causing severe anaemia. This range of responses depends on a patient’s other characteristics. For instance, how strong are their red cell membranes? How effective are their body systems in reducing inflammation?
Conventional treatments for those presenting with sickle cell crises have included admission, intravenous rehydration and pain relief if required. Antibiotics and good nursing too, along with careful nutrition and social supports have prolonged life and reduced hospitalisation in most sufferers. Blood transfusions are often given to increase the numbers of red cells. More recently, the agent voxelior has been introduced as it stops those “lumpy” or polymerisation changes in haemoglobin. Another medication, crizanlizumab, reduces “sticky” changes in blood vessels. New treatments currently prescribed in some centres include the amino acid L-glutamine, or mitapivat, both of which reduce the breakdown of red cells and therefore the severity of any crises.
A “curative” treatment for sickle cell can be the transplantation of stem cells. This requires a donor with the same tissue type as the patient: their stem cells may be used to repopulate the bone marrow of a sickle cell patient whose own stem cells have been removed. Optimal donors of stem cells are one’s siblings. Unrelated but tissue-matched donors can be used, but complication rates of such transplants are greater. Finding matches is challenging for many patients, so the drives to find other possible “cures” are strong.
Most patients with sickle cell anaemia get great benefit from increasing their foetal haemoglobin. This type of haemoglobin is particularly good at carrying oxygen – we use it as a foetus to collect oxygen from our mother’s placenta. After birth, the foetal haemoglobin gene is usually turned down to a low level, or switched off. Treatment with hydroxyurea can increase foetal haemoglobin pharmacologically, so this treatment is introduced relatively early to those with troubling sickle cell. But it is tempting to devise a genetic method to turning the gene back on to increase the amount of foetal haemoglobin. Alternatively, is it possible to consider repairing the original genetic defect that causes the sickle cell condition in the first place?
A number of different, precise editing methods allow the DNA that makes up our genes to be repaired. These processes take place day to day, as thousands of defects develop in our genome. Correction is necessary before cells divide. Repairing enzymes may be employed to fine-tune changes to small areas of the genome. A great challenge if this is used as a treatment is to ensure that cells with repaired genes multiply and take over (or repopulate) from any defective predecessors. The fidelity of any genetic repair process used as a treatment needs careful monitoring. There can be no tolerance for “off-target damage”.
Several trials of genetic repair strategies for sickle cell are in progress. Reassuring results from the Ruby Trial were reported last month. This early study involved a small group of patients with severe sickle cell disease. Some of their haematopoietic stem cells were collected and edited in a laboratory in order to increase foetal haemoglobin production. The patients were given medication to remove their remaining stem cells. Edited cells were then returned to them. All those in the trial did well; their edited cells repopulated. Patients showed increased levels of foetal haemoglobin. No complications were seen. This proof of concept trial showed that gene editing might be developed to manage sickle cell anaemia. Although too early to claim this was curative, it is a crucial first step.
Our neighbours have been reassured. Father and daughter – but not mother – have sickle cell trait. Testing one’s DNA can reveal useful, if uncomfortable genetic information! Breaking anonymity in this way can cause some familial chaos. However, when it comes to correcting sickle cell anaemia, one’s family links trump anonymity.
Useful resources:
https://www.nhs.uk/conditions/sickle-cell-disease/
https://www.cdc.gov/ncbddd/sicklecell/index.html
Dr. Colin Michie is currently the Associate Dean for Research and Knowledge Exchange at the School of Medicine in the University of Central Lancashire. He specializes in paediatrics, nutrition, and immunology. Michie has worked in the UK, southern Africa and Gaza as a paediatrician and educator and was the associate Academic Dean for the American University of the Caribbean Medical School in Sint Maarten a few years ago.



